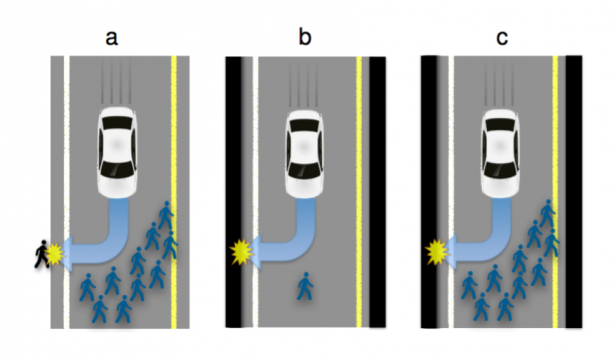
The wide adoption of self-driving, Autonomous Vehicles (AVs) promises to dramatically reduce the number of traffic accidents. Some accidents, though, will be inevitable, because some situations will require AVs to choose the lesser of two evils. For example, running over a pedestrian on the road or a passer-by on the side; or choosing whether to run over a group of pedestrians or to sacrifice the passenger by driving into a wall. It is a formidable challenge to define the algorithms that will guide AVs confronted with such moral dilemmas. In particular, these moral algorithms will need to accomplish three potentially incompatible objectives: being consistent, not causing public outrage, and not discouraging buyers. We argue to achieve these objectives, manufacturers and regulators will need psychologists to apply the methods of experimental ethics to situations involving AVs and unavoidable harm. To illustrate our claim, we report three surveys showing that laypersons are relatively comfortable with utilitarian AVs, programmed to minimize the death toll in case of unavoidable harm. We give special attention to whether an AV should save lives by sacrificing its owner, and provide insights into (i) the perceived morality of this self-sacrifice, (ii) the willingness to see this self-sacrifice being legally enforced, (iii) the expectations that AVs will be programmed to self-sacrifice, and (iv) the willingness to buy self-sacrificing AVs.
Yazar: Ferhat Kurt
Ankara Deep Learning – Derin Öğrenme Etkinliği
13 Ekim 2015 saat 19:00’da Hacettepe Teknokent Safir Bloklar Konferans Salonunda gerçekleştirdiğimiz Derin Öğrenme etkinliğinin video ve sunum dosyasına aşağıdan erişebilirsiniz.
Sunum Videosu
Sunum
![]() derin_ogrenme_linked_13.10.2015.pdf
derin_ogrenme_linked_13.10.2015.pdf
Etkinlikten Resimler
















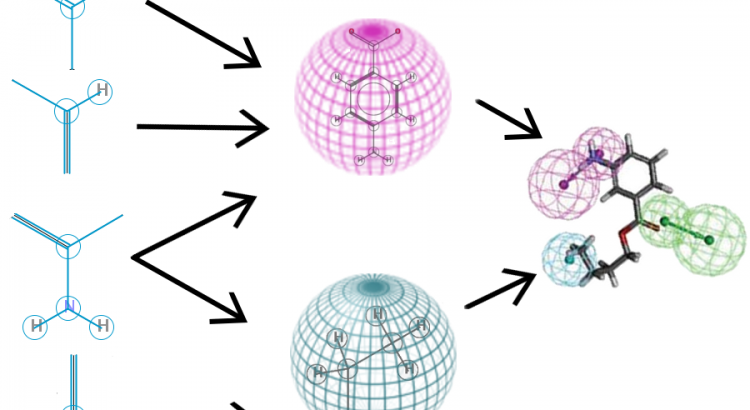
Makale: Deep Learning as an Opportunity in Virtual Screening
Deep learning excels in vision and speech applications where it pushed the stateof-the-art to a new level. However its impact on other fields remains to be shown. The Merck Kaggle challenge on chemical compound activity was won by Hinton’s group with deep networks. This indicates the high potential of deep learning in drug design and attracted the attention of big pharma. However, the unrealistically small scale of the Kaggle dataset does not allow to assess the value of deep learning in drug target prediction if applied to in-house data of pharmaceutical companies. Even a publicly available drug activity data base like ChEMBL is magnitudes larger than the Kaggle dataset. ChEMBL has 13 M compound descriptors, 1.3 M compounds, and 5 k drug targets, compared to the Kaggle dataset with 11 k descriptors, 164 k compounds, and 15 drug targets.
On the ChEMBL database, we compared the performance of deep learning to seven target prediction methods, including two commercial predictors, three predictors deployed by pharma, and machine learning methods that we could scale to this dataset. Deep learning outperformed all other methods with respect to the area under ROC curve and was significantly better than all commercial products. Deep learning surpassed the threshold to make virtual compound screening possible and has the potential to become a standard tool in industrial drug design.
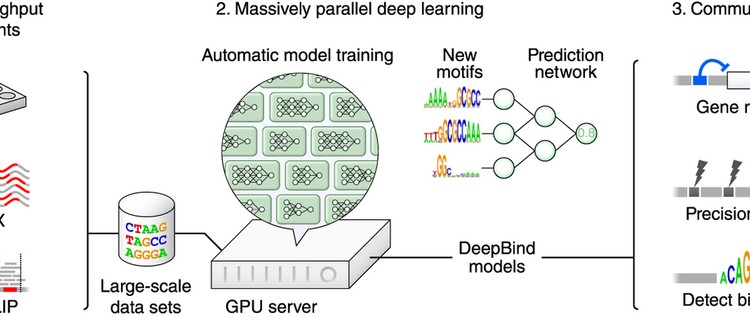
Makale: Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning
Knowing the sequence specificities of DNA- and RNA-binding proteins is essential for developing models of the regulatory processes in biological systems and for identifying causal disease variants. Here we show that sequence specificities can be ascertained from experimental data with ‘deep learning’ techniques, which offer a scalable, flexible and unified computational approach for pattern discovery. Using a diverse array of experimental data and evaluation metrics, we find that deep learning outperforms other state-of-the-art methods, even when training on in vitro data and testing on in vivo data. We call this approach DeepBind and have built a stand-alone software tool that is fully automatic and handles millions of sequences per experiment. Specificities determined by DeepBind are readily visualized as a weighted ensemble of position weight matrices or as a ‘mutation map’ that indicates how variations affect binding within a specific sequence.
Makale: Massively Multitask Networks for Drug Discovery
Massively multitask neural architectures provide a learning framework for drug discovery that synthesizes information from many distinct biological sources. To train these architectures at scale, we gather large amounts of data from public sources to create a dataset of nearly 40 million measurements across more than 200 biological targets. We investigate several aspects of the multitask framework by performing a series of empirical studies and obtain some interesting results: (1) massively multitask networks obtain predictive accuracies significantly better than single-task methods, (2) the predictive power of multitask networks improves as additional tasks and data are added, (3) the total amount of data and the total number of tasks both contribute significantly to multitask improvement, and (4) multitask networks afford limited transferability to tasks not in the training set. Our results underscore the need for greater data sharing and further algorithmic innovation to accelerate the drug discovery process.